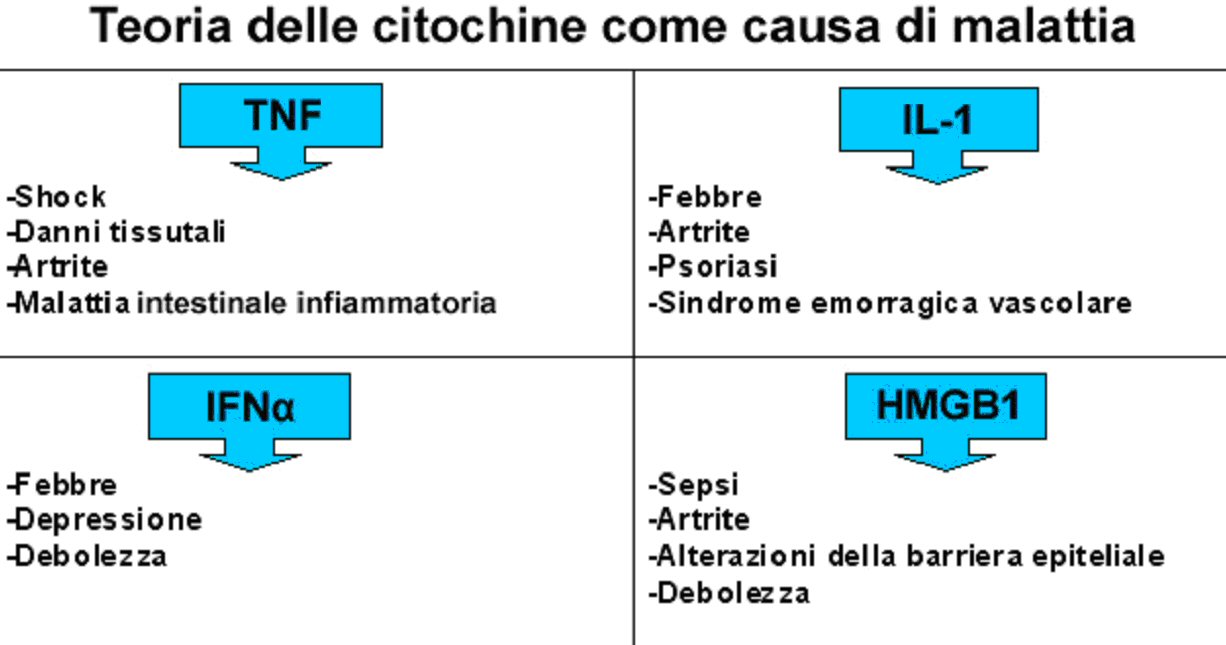
2. IL RIFLESSO INFIAMMATORIO
Il sistema nervoso è composto di sistemi sensoriali (che rilevano lo stato del corpo e degli organi) e di sistemi motori (che trasmettono segnali al corpo e agli organi). Il sistema motorio somatico controlla i movimenti volontari, mentre il sistema motorio autonomo controlla involontariamente le funzioni viscerali. Le due parti del sistema nervoso automo – il parasimpatico e il simpatico – cregolano di continuo le risposte fisiologiche essenziali, come la frequenza cardiaca e la pressione sanguigna, la frequenza respiratoria, la motilità gastro-intestinale, e la temperatura corporea.
Il sistema nervoso autonomo riceve informazioni da recettori sensoriali specializzati per rilevare lo stato fisiologico, per esempio i barorecettori per monitorare la pressione sanguigna, i recettori di stiramento e i chemocettori per monitorare l’attività gastrica. Il cervello primitivo, incluso il sistema limbico (che assolve a importanti funzioni della memoria), il tronco encefalico e l’ipotalamo ricevono segnali da questi recettori, e coordinano le risposte neurali del simpatico e del parasimpatico per mantenere l’omeostasi cardiovascolare o per regolare la digestione.
Queste funzioni autonome sono in genere subconscie, perché l’ipotalamo agisce come un portiere, lasciando penetrare fino ai centri cerebrali più alti <1%? delle informazioni ascendenti. Comunque, le funzioni vitali essenziali autonome possono essere modulate da segnali che nascono nel cervello superiore (corteccia cerebrale). Per esempio, si possono allenare dei soggetti con il bio-feedback in modo che riducano la frequenza cardiaca attraverso l’aumento dell’attività delle fibre efferenti del parasimpatico.
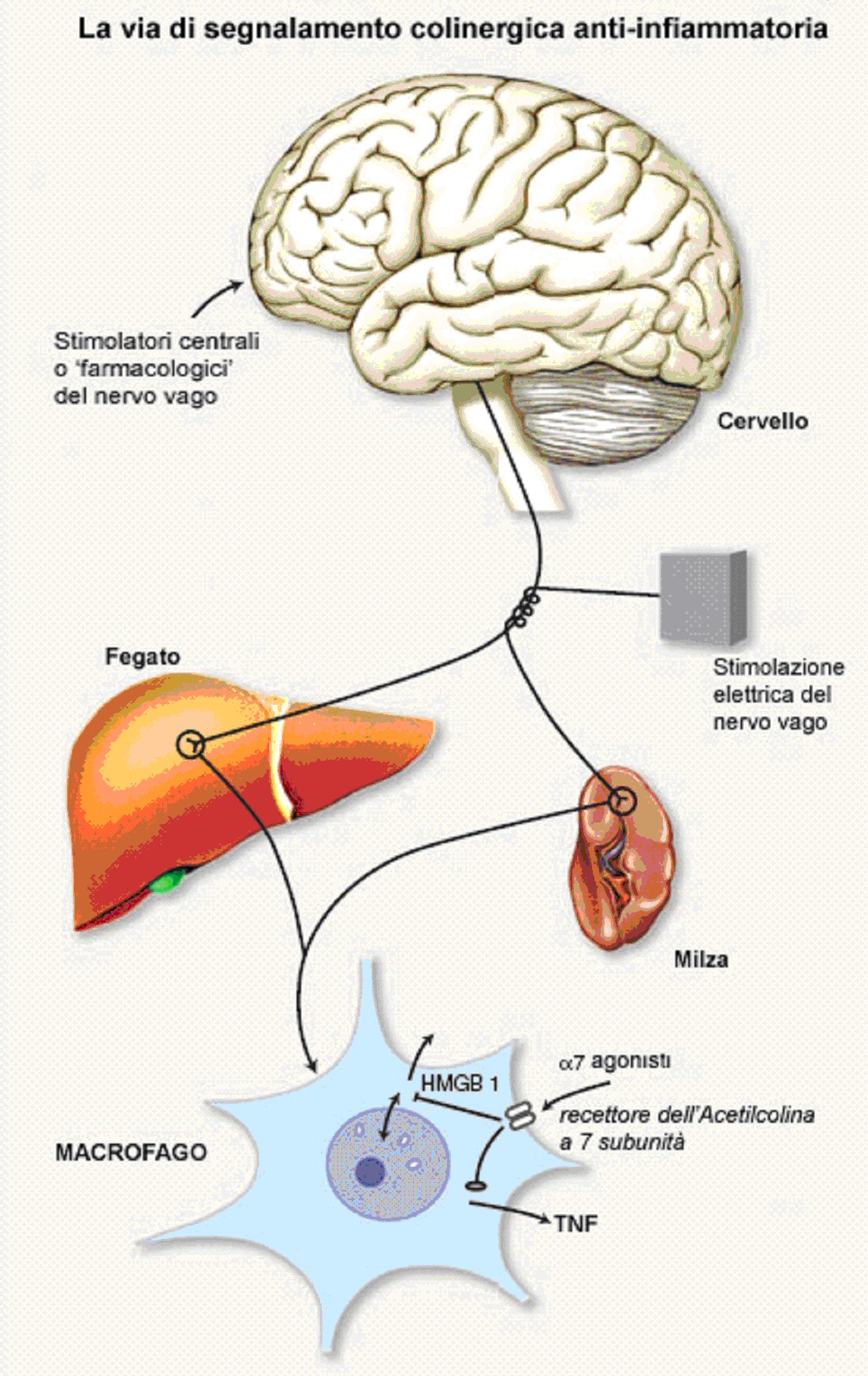
Nuovi studi hanno rivelato una via neurale essenziale che automaticamente controlla e modula la risposta infiammatoria (Fig. 2). Gli stimoli infiammatori attivano all’interno del nervo vago vie di segnalamento afferenti che sono poi ritrasmesse all’ipotalamo. Come il baroriflesso, in cui lo stiramento della carotide o dei seni aortici sollecitano una risposta efferente per rallentare il cuore e indurre una vasodilatazione periferica, l’input infiammatorio attiva una risposta anti-infiammatoria che previene un’eccessiva produzione di citochine. Il sistema nervoso integra la risposta infiammatoria. Esso raccoglie informazioni sugli eventi infiammatori o invasivi da diversi siti, mobilita le difese, e crea una memoria per migliorare le chance di sopravvivenza.
Rilevazione dell’infiammazione periferica
Il sistema nervoso centrale (CNS) riceve un input dal sistema immunitario sia attraverso vie umorali che vie neurali. Blalock ha suggerito che il sistema immunitario funziona come un “sesto senso” che rileva le invasioni microbiche e produce molecole per trasmettere queste informazioni al cervello [1, 2]. IL-1Alfa, TNF e altri mediatori immunologici possono avere accesso a centri del cervello, noti come regioni circumventricolari, che sono privi di barriera sanguigna cerebrale, o possono penetrare in altre aree per mezzo di sistemi di trasporto attivi. Ma IL-1Beta e l’endotossina possono anche attivare segnali afferenti del nervo vago, portando alla manifestazione di sintomi di malattia, inclusa l’avversione per il cibo e la febbre. Sia le vie umorali che quelle neurali che dal sistema immunitario vanno alla comunicazione col sistema nervoso sono state ritenute coinvolte nello sviluppo di febbre, anoressia, attivazione delle risposte ipotalamo-ipofisarie contro infezioni e danni, e nell’origine di altre manifestazioni comportamentali di malattia.
Meccanismi umorali della comunicazione immuno-cerebrale
Il fattore di necrosi tumorale e l’IL-Beta sono attivamente trasportati attraverso la barriera sanguigna cerebrale, e l’IL-1Alfa entra direttamente nel cervello attraverso gli organi circumventricolari come la ghiandola pineale, l’area postrema, l’eminenza mediana e il lobo neurale dell’ipofisi, dove la barriera sanguigna cerebrale è inesistente o discontinua [3-9]. L’IL-1Alfa penetra anche in queste aree del cervello attraverso un ipotetico meccanismo di trasporto a livello dei vasi [8]. La microglia e le cellule endoteliali della vascolarizzazione del cervello esprimono i recettori IL-1Beta, insieme ai co-recettori per l’endotossina, CD14 e al recettore-toll 4 [10-15]. Una volta che le citochine hanno accesso agli organi circumventricolari, esse possono produrre effetti a vasta portata attraverso secondi messaggeri come la ciclossigenasi -2, che induce l’acido arachidonico a rilasciare la prostaglandina E2 (PGE2); i recettori PG si localizzano in aree cerebrali che giocano ruoli importanti nelle risposte infiammatorie periferiche, come l’attivazione dell’asse ipotalamo-ipofisi-surrene e della febbre [16-18]. I mediatori infiammatori originati nel sangue, di alto peso molecolare, possono perciò informare direttamente il sistema nervoso centrale delle condizioni infiammatorie delle aree periferiche. La via umorale sembra essere un mezzo predominante per la comunicazione tra sistema immunitario e cervello quando i livelli di citochine circolanti sono alti.
Meccanismi neurali di comunicazione immuno-cerebrale
Anche quando i livelli di citochine circolanti sono bassi, il sistema nervoso centrale viene informato sullo stato dell’infiammazione attraverso segnali neurali afferenti. L’innervazione sensoriale degli organi immunitari per mezzo di fibre ascendenti che viaggiano nel nervo vago, come anche attraverso altre vie sensoriali del dolore ascendenti, fornisce un input importante sullo stato degli attacchi invasivi e dannosi in diversi compartimenti disseminati nel corpo [19]. Fatto importante, queste vie neurali che avvertono l’infiammazione funzionano secondo soglie più basse di rilevamento, e attivano risposte anche quando gli agenti infiammatori sono presenti nei tessuti a livelli che non sono abbastanza alti da raggiungere il cervello attraverso la circolazione del sangue. Il nervo vago è posizionato particolarmente bene per trasmettere informazioni tra il sistema immunitario e il sistema nervoso centrale, poiché esso innerva, tra gli altri organi, il fegato, i polmoni, i reni, il tratto digestivo, e altri organi viscerali che agiscono sia come filtri per i patogeni e i loro prodotti, sia come vie d’ingresso per i patogeni stessi. Watkins e colleghi hanno fornito uno studio critico sul ruolo sensoriale delle fibre afferenti del nervo vago, osservando che la vagotomia diminuisce l’innalzamento della febbre in animali esposti a basse dosi intra-addominali di IL-1 o di endotossine [20-23]. Non è del tutto chiaro come il nervo vago “rilevi” la presenza dei mediatori infiammatori, ma i neuroni nel nervo vago esprimono l’ mRNA del recettore per l’IL-1, e alcuni siti leganti l’IL-1 sono stati identificati, nel nervo vago vero e proprio e sulle cellule glomerulari [24,25]. Studi elettro-fisiologici indicano che i segnali del nervo vago possono essere attivati anche da meccano-recettori, chemocettori, sensori della temperatura e sensori di osmolarità che possono essere attivati in un sito infiammatorio [26].
Integrazione dei segnali infiammatori periferici
La rilevazione dell’infiammazione periferica attiva segnali afferenti umorali e neurali che convergono sul tronco encefalico, dove i segnali vengono esaminati, e dove vengono coordinate appropriate risposte neurali e umorali. La maggior parte delle informazioni ottenute dal sistema di percezione sensoriale vengono organizzate in modo somato-topico, così che l’input sensoriale da un distinto luogo periferico venga precisamente localizzato in una via di fibre ascendenti e nel cervello. Il modello fatto ad arco dell’homunculus sensoriale si può applicare alla sensazione somatica come pure alla percezione sensoriale conscia. E’ ora ragionevole suggerire che ci può essere anche un ordine somatotopico dell’input sensoriale da parte del sistema immunitario. Le fibre afferenti del nervo vago terminano nell’area postrema e nel nucleo del tratto solitario, due regioni all’interno del cervello che sono particolarmente attive durante l’infiammazione periferica [18, 27]. I segnali del nervo vago ascendente sollecitano neuroni di secondo ordine all’interno del nucleo del tratto solitario (NTS) attraverso l’attività del glutammato [28]. Il nucleo del tratto solitario forma l’apice di un ciclo di controllo vago-vagale che modula l’attività viscerale regolata vagalmente attraverso due meccanismi distinti. Prima, i neuroni NTS inibiscono un subset di neuroni all’interno del nucleo motore dorsale del vago (DMV) che fornisce segnali eccitatori colinergici agli organi interni, incluso il tratto digestivo [29]. In un secondo tempo, il nucleo del tratto solitario sopprime anche l’attività viscerale, mettendo in funzione altri neuroni inibitori efferenti DMV attraverso vie metaboliche non-adrenalinergiche e non-colinergiche [30]. Fatto importante, sia NTS che DMV contengono vasi sanguigni che sono privi di una barriera cerebrale sanguigna, cosa che rende questi sistemi importanti organi circumventricolari. Questo può permettere sia al nucleo del tratto solitario che al nucleo motore dorsale di ricevere un input sensoriale da parte di fattori circolanti diffusibili come il lipopolisaccaride, il TNF, o IL-1, oltre ai segnali del nervo vago afferente [29, 31]. La segnalazione colinergica attraverso i recettori muscarinici centrali sembra essere una componente decisiva delle vie metaboliche che regolano la segnalazione efferente attraverso la via colinergica anti-infiammatoria, perché la muscarina somministrata nel cervello dei ratti diminuisce significativamente le risposte infiammatorie alla stimolazione con endotossina periferica, mentre la muscarina somministrata nelle aree periferiche non ha effetto sull’infiammazione periferica (V. A. Pavlov and K. J: Tracey, data non pubblicata). La comunicazione tra NTS, DMV, ipotalamo, e forse le funzioni cerebrali più alte, coordina le risposte umorali e neurali che modulano la funzione del sistema immunitario.
Regolazione colinergica dell’infiammazione periferica
Abbiamo recentemente scoperto che i neuroni colinergici possono regolare la sintesi del fattore di necrosi tumorale attraverso l’acetilcolina [32, 33]. Questa è definita la “via anti-infiammatoria colinergica” e poiché l’ACh è il principale neurotrasmettitore para-simpatico, i macrofagi esposti ad ACh vengono effettivamente disattivati. Il nervo vago (che è così chiamato per il suo percorso vagante) innerva gli organi maggiori, inclusi quelli che ospitano il sistema reticolo-endoteliale (fegato, polmoni, milza, reni e intestino) [34]. L’attivazione sperimentale della via colinergica anti-infiammatoria con la stimolazione elettrica diretta delle fibre efferenti del nervo vago inibisce la sintesi di TNF nel fegato, nella milza e nel cuore, ed attenua i livelli di TNF nel siero durante l’infezione da endotossine, il danno da ischemia/riperfusione, lo shock emorragico, ed altre sindromi associate con il rilascio eccessivo di citochine [32, 33, 35-38]. La vagotomia esagera le risposte del TNF agli stimoli infiammatori e sensibilizza gli animali agli effetti letali dell’endotossina; ciò suggerisce che i segnali colinergici anti-infiammatori trasmessi attraverso il nervo vago efferente giocano un ruolo nel mantenimento dell’omeostasi immunitaria [32, 35].
La sinapsi tra il sistema nervoso colinergico e il sistema immunitario naturale ha bisogno di un nicotinico, il recettore Alfa-bungarotossina-sensibile dell’ACh espresso sui macrofagi e su altre cellule immunocompetenti che modulano o partecipano alla risposta infiammatoria [33]. Gli agonisti colinergici, inclusa la nicotina e l’ACh, inibiscono significativamente il rilascio di TNF e di altre citochine da parte di macrofagi umani stimolati da endotossina. I macrofagi dei tessuti, monociti non-circolanti, producono la maggior parte delle citochine infiammatorie rilasciate sistemicamente durante le risposte infiammatorie eccessive. L’interazione degli agonisti colinergici con i recettori per l’ACh inibisce la sintesi di citochine pro-infiammatorie, ma non di citochine anti-infiammatorie (per es. IL-10) [32, 33, 39]. In confronto ai macrofagi, i monociti sono refrattari agli effetti inibenti il rilascio di citochine da parte dell’ACh; solo concentrazioni oltre la soglia fisiologica di agonisti colinergici inibiscono la sintesi di citochine monocitarie [32].
L’espressione macrofagica della sub-unità Alfa7 del recettore nicotinico per l’ACh distingue la via anti-infiammatoria colinergica dall’attività dei recettori muscarinici identificati precedentemente sui linfociti, sulle cellule sanguigne mononucleari periferiche, e sui macrofagi alveolari [40, 41]. L’attivazione dei recettori macrofagici dell’ACh inibisce specificamente la segnalazione del NF-KB endotossina-indotto, ma non influenza l’attivazione di varie proteine-chinasi mitogeno-associate che sono tipicamente unite alla segnalazione dell’endotossina [39]. L’affermazione che i macrofagi sono altamente sensibili all’acetilcolina suggerisce che altre cellule non-neuronali che producono l’ACh (per es. le cellule epiteliali, i linfociti T, le cellule endoteliali) possono anche partecipare alla modulazione della funzione dei macrofagi situati nel tessuto adiacente [42, 43].
La maggior parte degli studi compiuti fino ad oggi, caratterizzanti la via colinergica-anti-infiammatoria, si sono focalizzati sull’interazione macrofago-ACh, ma anche altri tipi di cellule, in particolare quelle dell’endotelio, sono potenzialmente regolate dall’acetilcolina. L’attivazione delle cellule endoteliali durante l’infiammazione, caratterizzata da un’aumentata espressione di molecole di adesione e da una produzione di mediatori infiammatori, gioca un ruolo decisivo nell’adesione e nella conseguente trasmigrazione di leucociti infammatori. Mediatori infiammatori come le chemochine presenti sulla superficie dell’endotelio attivano progressivamente i leucociti che si muovono lungo la parete dei vasi; per esempio, i neutrofili vengono attivati attraverso il legame con molecole di adesione presenti sull’endotelio [44-46]. Le cellule microvascolari dell’endotelio umano esprimono la sub-unità Alfa7 del recettore per l’ACh sulla superficie delle cellule, e l’ACh blocca significativamente l’espressione delle molecole di adesione TNF-indotta e l’espressione delle chemochine in una maniera concentrazione-dipendente [47].
La segnalazione colinergica attraverso agonisti dei recettori (per es.la nicotina) o attraverso la stimolazione elettrica diretta del nervo vago inibiscono la trasmigrazione dei leucociti attraverso l’endotelio in vivo; fatto notevole, quest’effetto può essere bloccato con la mecamilamina, un modulatore allosterico negativo dei recettori nicotinici di ACh, cosa che indica che gli effetti degli agonisti colinergici non sono dovuti all’attività di interazioni con altri recettori [47]. ACh modula anche le risposte infiammatorie intestinali, e diminuisce il rilascio di istamina da parte delle mastocellule delle mucose delle vie aeree [48-50]. La nicotina ha un effetto anti-infiammatorio su molti altri tipi di cellule, inclusi i monociti, le cellule epiteliali, le cellule T e i neutrofili, ma il ruolo della sub-unità Alfa7 del recettore per l’ACh in queste cellule non è stato ancora dettagliatamente determinato [51-55]. |
3. IMPLICAZIONI TERAPEUTICHE DELLA VIA COLINERGICA ANTI-INFIAMMATORIA
I meccanismi fisiologici anti-infiammatori rappresentano dei sistemi efficienti che sono stati selezionati dall’evoluzione per controllare l’infiammazione. Essi possono anche essere sfruttati per il trattamento dei disordini infiammatori. Varie metodologie sono state adottate per gli animali quando si sono scoperte e descritte le vie colinergiche anti-infiammatorie che potrebbero essere subito adattate per uso terapeutico [32, 33, 39] (Fig. 3).
Stimolazione elettrica del nervo vago
La connessione “cablata” tra il sistema nervoso e quello immunitario funziona come un meccanismo anti-infiammatorio in vari modelli di infiammazione sistemica e locale. I ratti sottoposti a vagotomia cervicale bilaterale sono più sensibili a una successiva iniezione di endotossina degli animali che hanno subito un intervento di chirurgia placebo; gli animali vagotomizzati vanno più rapidamente soggetti a shock endo-tossiemico e mostrano una risposta infiammatoria più forte in confronto agli animali operati con chirurgia placebo. La stimolazione elettrica del nervo vago diminuisce il rilascio di citochine pro-infiammatorie, incluso il TNF, e di IL-1Beta nell’endo-tossiemia del topo, e protegge anche contro lo shock endo-tossiemico (ipotensione) [32, 33, 35]. La stimolazione del nervo vago ha effetti simili in vari modelli animali di malattie sistemiche infiammatorie, incluso la peritonite/sepsi, il danno da ischemia/riperfusione e lo shock ipovolemico [36, 37, osservazioni personali degli autori]. È’ interessante notare che la stimolazione elettrica del nervo vago diminuisce anche le risposte infiammatorie in modelli animali di infiammazione locale, incluso l’edema della zampa carragenina-indotto e il modello della sacca d’aria con carragenina; questi modelli d’infiammazione cutanea sono caratterizzati da un’elevata attivazione di cellule endoteliali [47, 56].
I parametri del voltaggio e della frequenza sufficienti ad attivare la via anti-infiammatoria colinergica sono sotto la soglia richiesta per attivare le fibre vagali del cuore, fatto che indica che la stimolazione del nervo vago può essere un mezzo pratico ed efficiente per regolare terapeuticamente l’infiammazione periferica nelle patologie umane (osservazioni personali degli autori)). Questo approccio alla regolazione dell’infiammazione sistemica o locale può avere un potenziale terapeutico significativo perché gli stimolatori del nervo vago sono strumenti clinicamente approvati, usati per l’epilessia e la depressione in pazienti refrattari ad altri trattamenti.
Gli agonisti della subunità Alfa7 : una nuova classe di agenti anti-infiammatori
Un altro approccio terapeutico per controllare la via colinergica anti-infiammatoria nelle malattie da infiammazione è quella di usare gli agonisti colinergici che attivano specificamente la sub-unità macrofagica Alfa7 del recettore per l’ACh. I modelli animali in cui si sono usati approcci farmacologici hanno fornito dei dati preliminari che indicano che tale tipo di metodologia potrebbe avere successo. La nicotina, un agonista della subunità Alfa7 relativamente non specifico, fornisce un notevole vantaggio in termini di sopravvivenza ad animali soggetti ad infezione da endotossina o a peritonite/sepsi, e blocca il decorso di coliti ulcerose o infiammazioni cutanee indotte sperimentalmente [39, 52, 57, 58]. Sebbene le proprietà anti-infiammatorie della nicotina siano ben descritte, il meccanismo preciso attraverso il quale essa inibisce l’infiammazione non è stato in passato compreso. La nicotina blocca la migrazione dei leucociti nelle sacche d’aria iniettate con carragenina, e diminuisce l‘infiltrato nelle orecchie dei topi soggetti alla reazione di Shwartzman. L’effetto della nicotina sul reclutamento della cellula infiammatoria dipende dai recettori colinergici, poiché la mecamilamina antagonista del recettore annulla l’effetto protettivo della nicotina [47].
L’agonista endogeno della subunità Alfa7, l’ACh, è un modulatore anti-infiammatorio fisiologico del rilascio di HMGB1. La via anti-infiammatoria colinergica può essere sfruttata per il trattamento di gravi sepsi [39]. La nicotina mima gli effetti inibitori dell’ACh sul rilascio di HMGB1, attraverso un meccanismo che richiede che la sub-unità Alfa7 del recettore ACh inibisca la via metabolica dell’NF-KB [39]. Nuovi e più specifici agonisti della subunità Alfa7 sono inoltre in corso di sviluppo e di studio. Gli autori hanno scoperto che gli agonisti della subunità Alfa7 sono efficaci inibitori della traslocazione nucleare di NF-KB, del rilascio di citochine e di cellule endoteliali e dell’attivazione di macrofagi, in modelli murini di infiammazione locale (sacca d’aria con carragenina) e sistemica (infezione con endotossina, peritonite/sepsi) [39, 47]. L’inibizione della via di NF-KB potrebbe non essere il meccanismo d’azione primario di questi agenti, e ulteriori meccanismi di trasduzione del segnale che portino alla disattivazione dei macrofagi sono in corso di studio riguardo all’effetto anti-infiammatorio di agonisti della subunità Alfa7 in altri modelli infiammatori.
L’uso dell’agonista colinergico, la nicotina, è stato trovato efficace in molti severi trial clinici. Trial randomizzati con uso di placebo hanno dimostrato che il trattamento con nicotina è stato associato a remissioni significative della gravità di coliti ulcerose [59-62]. Mentre il meccanismo preciso degli effetti anti-infiammatori della nicotina sulle malattie dell’uomo non è ancora stato provato, dati recenti ottenuti da colture cellulari e studi sugli animali indicano che la protezione può essere stata raggiunta con la diminuzione del rilascio delle citochine. |
5. IMPLICAZIONI E PROSPETTIVE
Gli effetti fisiologici dell’attività delle fibre efferenti del nervo vago classicamente descritti includono il rallentamento della frequenza cardiaca, la stimolazione della motilità gastrica, la dilatazione delle arteriole e il restringimento delle pupille. La regolazione della risposta infiammatoria si può ora aggiungere a questa lista. Una via di segnalamento anti-infiammatoria basata sui nervi fornisce molti vantaggi rispetto alle vie umorali che funzionano isolate. La rete anti-infiammatoria diffusa, che include i glucocorticoidi, le citochine anti-infammatorie, ed altri mediatori umorali, è di tipo concentrazione gradiente-dipendente, è lenta e non-integrata, così che i mediatori diffusibili possano raggiungere tutte le zone del corpo, incluse quelle senza risposte infiammatorie in corso [19]. Di contro, la via di segnalamento anti-infiammatoria colinergica è di tipo non continuo e localizzata in tessuti dove hanno tipicamente origine invasioni da microbi e danni. Confrontata con il ritmo biologico normale di una tipica, diffusibile risposta infiammatoria (ore a giorni), la segnalazione neurale è come un lampo veloce. La velocità di una via metabolica neurale fornisce segnali regolatori più capaci di contenere la risposta immunitaria precoce.
La descrizione della via anti-infiammatoria colinergica dimostra che il sistema nervoso centrale regola direttamente la risposta sistemica pro-infiammatoria delle citochine agli stimoli infiammatori. L’identificazione di questi meccanismi neurali endogeni ha diverse implicazioni terapeutiche per il trattamento delle condizioni infiammatorie. Si può provare che la stimolazione elettrica diretta del nervo vago ha un’attività anti-infiammatoria nell’uomo. Gli agonisti colinergici che agiscono localmente, come la nicotina, che legano direttamente i recettori dell’ACh sui macrofagi tessuto-residenti e su altre cellule del sistema immunitario naturale, possono rappresentare un altro mezzo per trarre vantaggio della via anti-infiammatoria colinergica allo scopo di regolare l‘infiammazione. Per esempio, la nicotina è stata usata per trattare efficacemente pazienti con colite ulcerosa, una condizione patologica associata alla produzione eccessiva di citochine pro-infiammatorie nella mucosa gastro-intestinale. Sebbene il meccanismo d’azione della nicotina nella colite ulcerosa sia sconosciuto, è interessante considerare il ruolo potenziale dei recettori colinergici simili al recettore nicotinico nel trattamento di questa patologia.
Gli animali con insufficiente segnalazione efferente nella via colinergica anti-infiammatoria mostrano forti risposte sistemiche alla malattia anti-infiammatoria suggerendo che questa via neurale fornisce un’energica regolazione dell’omeostasi immunitaria. Clinicamente, la disfunzione del sistema autonomo è stata associata con malattie infiammatorie come l’artrite reumatoide, il morbo di Crohn, il lupus, il diabete e la sepsi; se questa disfunzione tragga origine dalla componente infiammatoria di queste patologie, o ne sia in realtà una causa sottostante, è ancora poco chiaro. In una vasta sperimentazione clinica, il tasso di letalità nei pazienti con sepsi in condizioni mentali non normali è risultato doppio rispetto a quello di pazienti con attività mentale normale; la variabilità del rallentamento della frequenza cardiaca, un riflesso della disfunzione del sistema nervoso autonomo, è anche associata con l’aumento della mortalità per sepsi [72-75].
La scoperta che la via anti-infiammatoria colinergica rappresenta una potente via endogena, conservata durante l’evoluzione, con lo scopo di proteggere l’ospite dalla sua stessa risposta immunitaria, è un interessante nuovo sviluppo nell’immunologia clinica e di base. E’ probabile che, mentre apprendiamo più elementi sull’attività di questa via, diventerà possibile controllarla terapeuticamente, usando sia gli stimolatori del nervo vago, gli agonisti della subunità Alfa7, gli altri agenti in grado di aumentare la segnalazione del vago, sia il bio-feedback, tutto allo scopo di curare le malattie da produzione eccessiva di citochine.
Dichiarazione di conflitto d’interessi
Gli autori sono consulenti di Critical Therapeutics, Inc, una compagnia che sta sviluppando tecnologie riguardanti la via anti-infiammatoria colinergica. |
Note Bibliografiche
1) Blalock JE. The immune system as a sensory organ. J Immunol 1984; 132: 1067–70.
2) Blalock JE. Shared ligands and receptors as a molecular mechanism for communication between the immune and neuroendocrine systems. Ann N Y Acad Sci 1994; 741: 292–8.
3) Gross PM, Weindl A. Peering through the windows of the brain. J Cereb Blood Flow Metab 1987; 7: 663–72.
4) Banks WA, Kastin AJ. Blood to brain transport of interleukin links the immune and central nervous systems. Life Sci 1991; 48: PL117–21.
5) Gutierrez EG, Banks WA, Kastin AJ. Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J Neuroimmunol 1993; 47: 169–76.
6) Johnson AK, Gross PM. Sensory circumventricular organs and brain homeostatic pathways. FASEB J 1993; 7: 678–86.
7) Gutierrez EG, Banks WA, Kastin AJ. Blood-borne interleukin-1 receptor antagonist crosses the blood-brain barrier. J Neuroimmunol 1994; 55: 153–60.
8) Plotkin SR, Banks WA, Kastin AJ. Comparison of saturable transport and extracellular pathways in the passage of interleukin-1 alpha across the blood-brain barrier. J Neuroimmunol 1996; 67: 41–7.
9) Pan W, Banks WA, Kastin AJ. Blood-brain barrier permeability to ebiratide and TNF in acute spinal cord injury. Exp Neurol 1997; 146: 367–73.
10) Ericsson A, Liu C, Hart RP, Sawchenko PE. Type 1 interleukin-1 receptor in the rat brain: distribution, regulation, and relationship to sites of IL-1-induced cellular activation. J Comp Neurol 1995; 361: 681–98.
11) Wong ML, Licinio J. Localization of interleukin 1 type I receptor mRNA in rat brain. Neuroimmunomodulation 1994; 1: 110–5.
12) Yabuuchi K, Minami M, Katsumata S, Satoh M. Localization of type I interleukin-1 receptor mRNA in the rat brain. Brain Res Mol Brain Res 1994; 27: 27–36.
13) Cunningham ET, Jr, Wada E, Carter DB, Tracey DE, Battey JF, De Souza EB. In situ histochemical localization of type I interleukin-1 receptor messenger RNA in the central nervous system, pituitary, and adrenal gland of the mouse. J Neurosci 1992; 12: 1101–14.
14) Lacroix S, Rivest S. Effect of acute systemic inflammatory response and cytokines on the transcription of the genes encoding cyclooxygenase enzymes (COX-1 and COX-2) in the rat brain. J Neurochem 1998; 70: 452–66.
15) Laflamme N, Rivest S. Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J 2001; 15: 155–63.
16) Breder CD, Smith WL, Raz A et al. Distribution and characterization of cyclooxygenase immunoreactivity in the ovine brain. J Comp Neurol 1992; 322: 409–38.
17) Cao C, Matsumura K, Yamagata K, Watanabe Y. Endothelial cells of the rat brain vasculature express cyclooxygenase-2 mRNA in response to systemic interleukin-1 beta: a possible site of prostaglandin synthesis responsible for fever. Brain Res 1996; 733: 263–72.
18) Ericsson A, Arias C, Sawchenko PE. Evidence for an intramedullary prostaglandin-dependent mechanism in the activation of stress-related neuroendocrine circuitry by intravenous interleukin-1. J Neurosci 1997; 17: 7166–79.
19) Tracey KJ. The inflammatory reflex. Nature 2002; 420: 853–9.
20) Watkins LR, Maier SF. Implications of immune-to-brain communication for sickness and pain. Proc Natl Acad Sci U S A 1999; 96: 7710–3.
21) Watkins LR, Goehler LE, Relton JK et al. Blockade of interleukin- 1 induced hyperthermia by subdiaphragmatic vagotomy: evidence for vagal mediation of immune-brain communication. Neurosci Lett 1995; 183: 27–31.
22) Hansen MK, Daniels S, Goehler LE, Gaykema RP, Maier SF, Watkins LR. Subdiaphragmatic vagotomy does not block intraperitoneal
lipopolysaccharide-induced fever. Auton Neurosci 2000; 85: 83–7.
23) Hansen MK, O’Connor KA, Goehler LE, Watkins LR, Maier SF. The contribution of the vagus nerve in interleukin-1beta-induced fever is dependent on dose. Am J Physiol Regul Integr Comp Physiol 2001; 280: R929–34.
24) Goehler LE, Gaykema RP, Hansen MK, Anderson K, Maier SF, Watkins LR. Vagal immune-to-brain communication: a visceral chemosensory pathway. Auton Neurosci 2000; 85: 49–59.
25) Goehler LE, Relton JK, Dripps D et al. Vagal paraganglia bind biotinylated interleukin-1 receptor antagonist: a possible mechanism for immune-to-brain communication. Brain Res Bull 1997; 43: 357–64.
26) Berthoud HR, Neuhuber WL. Functional and chemical anatomy of the afferent vagal system. Auton Neurosci 2000; 85: 1–17.
27) Lee HY, Whiteside MB, Herkenham M. Area postrema removal abolishes stimulatory effects of intravenous interleukin- 1beta on hypothalamic-pituitary-adrenal axis activity and c-fos mRNA in the hypothalamic paraventricular nucleus. Brain Res Bull 1998; 46: 495–503.
28) Smith BN, Dou P, Barber WD, Dudek FE. Vagally evoked synaptic currents in the immature rat nucleus tractus solitarii in an intact in vitro preparation. J Physiol 1998; 512 (Pt 1): 149–62.
29) Rogers RC, McTigue DM, Hermann GE. Vagal control of digestion: modulation by central neural and peripheral endocrine factors. Neurosci Biobehav Rev 1996; 20: 57–66.
30) Rogers RC, Hermann GE, Travagli RA. Brainstem pathways responsible for oesophageal control of gastric motility and tone in the rat. J Physiol 1999; 514 (Pt 2): 369–83.
31) Broadwell RD, Sofroniew MV. Serum proteins bypass the blood-brain fluid barriers for extracellular entry to the central nervous system. Exp Neurol 1993; 120: 245–63.
32) Borovikova LV, Ivanova S, Zhang M et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000; 405: 458–62.
33) Wang H, Yu M, Ochani M et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003; 421: 384–8.
34) Bellinger DL, Lorton D, Lubahn C, Felten DL. In: Ader R, Felten DL, Cohen N, eds. Psychoneuroimmunology. San Diego, CA: Academic Press, 2001; 55–112.
35) Bernik TR, Friedman SG, Ochani M et al. Pharmacological stimulation of the cholinergic antiinflammatory pathway. J Exp Med 2002; 195: 781–8.
36) TR, Friedman SG, Ochani M et al. Cholinergic antiinflammatory pathway inhibition of tumor necrosis factor during ischemia reperfusion. J Vasc Surg 2002; 36: 1231–6.
37) Guarini S, Altavilla D, Cainazzo MM et al. Efferent vagal fibre stimulation blunts nuclear factor-kappaB activation and protects against hypovolemic hemorrhagic shock. Circulation 2003; 107: 1189–94.
38) Guarini S, Cainazzo MM, Giuliani D et al. Adrenocorticotropin reverses hemorrhagic shock in anesthetized rats through the rapid activation of a vagal anti-inflammatory pathway. Cardiovasc Res 2004; 63: 357–65.
39) Wang H, Liao H, Ochani M et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 2004; 10: 1216–21.
40) Sato E, Koyama S, Okubo Y, Kubo K, Sekiguchi M. Acetylcholine stimulates alveolar macrophages to release inflammatory cell chemotactic activity. Am J Physiol 1998; 274 (Pt 1): L970–9.
41) Sato KZ, Fujii T, Watanabe Y et al. Diversity of mRNA expression for muscarinic acetylcholine receptor subtypes and neuronal nicotinic acetylcholine receptor subunits in human mononuclear leukocytes and leukemic cell lines. Neurosci Lett 1999; 266: 17–20.
42) Wessler I, Kirkpatrick CJ, Racke K. Non-neuronal acetylcholine, a locally acting molecule, widely distributed in biological systems: expression and function in humans. Pharmacol Ther 1998; 77: 59–79.
43) Kawashima K, Fujii T. Extraneuronal cholinergic system in lymphocytes. Pharmacol Ther 2000; 86: 29–48.
44) Ley K. Integration of inflammatory signals by rolling neutrophils. Immunol Rev 2002; 186: 8–18.
45) Simon SI, Hu Y, Vestweber D, Smith CW. Neutrophil tethering on E-selectin activates beta 2 integrin binding to ICAM-1 through a mitogen-activated protein kinase signal transduction pathway. J Immunol 2000; 164: 4348–58.
46) Hidari KI, Weyrich AS, Zimmerman GA, McEver RP. Engagement of P-selectin glycoprotein ligand-1 enhances tyrosine phosphorylation and activates mitogen-activated protein kinases in human neutrophils. J Biol Chem 1997; 272: 28750–6.
47) Saeed RW, Varma S, Peng T et al. Cholinergic stimulation blocks leukocyte recruitment during inflammation: endothelial cell-mediated effects. 2005, J Exp Med, in press.
48) Davis KA, Masella J, Blennerhassett MG. Acetylcholine metabolism in the inflamed rat intestine. Exp Neurol 1998; 152: 251–8.
49) Reinheimer T, Mohlig T, Zimmermann S, Hohle KD, Wessler I. Muscarinic control of histamine release from airways. Inhibitory M1-receptors in human bronchi but absence in rat trachea. Am J Respir Crit Care Med 2000; 162 (Pt 1): 534–8.
50) Reinheimer T, Baumgartner D, Hohle KD, Racke K, Wessler I. Acetylcholine via muscarinic receptors inhibits histamine release from human isolated bronchi. Am J Respir Crit Care Med 1997; 156 (Pt 1): 389–95.
51) Middlebrook AJ, Martina C, Chang Y, Lukas RJ, DeLuca D. Effects of nicotine exposure on T cell development in fetal thymus organ culture: arrest of T cell maturation. J Immunol 2002; 169: 2915–24.
52) Sykes AP, Brampton C, Klee S, Chander CL, Whelan C, Parsons ME. An investigation into the effect and mechanisms of action of nicotine in inflammatory bowel disease. Inflamm Res 2000; 49: 311–9.
53) Summers AE, Whelan CJ, Parsons ME. Nicotinic acetylcholine receptor subunits and receptor activity in the epithelial cell line HT29. Life Sci 2003; 72: 2091–4.
54) Sopori ML, Kozak W, Savage SM et al. Effect of nicotine on the immune system: possible regulation of immune responses by central and peripheral mechanisms. Psychoneuroendocrinology 1998; 23: 189–204.
55) Pabst MJ, Pabst KM, Collier JA et al. Inhibition of neutrophil and monocyte defensive functions by nicotine. J Periodontol 1995; 66: 1047–55.
56) Borovikova LV, Ivanova S, Nardi D et al. Role of vagus nerve signaling in CNI-1493-mediated suppression of acute inflammation. Auton Neurosci 2000; 85: 141–7.
57) Eliakim R, Karmeli F. Divergent effects of nicotine administration on cytokine levels in rat small bowel mucosa, colonic mucosa, and blood. Isr Med Assoc J 2003; 5: 178–80.
58) Sopori ML, Kozak W, Savage SM, Geng Y, Kluger MJ. Nicotine- induced modulation of T Cell function. Implications for inflammation and infection. Adv Exp Med Biol 1998; 437: 279–89.
59) Jick H, Walker AM. Cigarette smoking and ulcerative colitis. N Engl J Med 1983; 308: 261–3.
60) Rampton DS. Smoking and ulcerative colitis. Lancet 1984; 1: 168.
61) Pullan RD, Rhodes J, Ganesh S et al. Transdermal nicotine for active ulcerative colitis. N Engl J Med 1994; 330: 811–5.
62) Ando Y. Transdermal nicotine for ulcerative colitis. Ann Intern Med 1997; 127: 491–2.
63) Toussirot E, Serratrice G, Valentin P. Autonomic nervous system involvement in rheumatoid arthritis. 50 cases. J Rheumatol 1993; 20: 1508–14.
64) Tan J, Akin S, Beyazova M, Sepici V, Tan E. Sympathetic skin response and R-R interval variation in rheumatoid arthritis. Two simple tests for the assessment of autonomic function. Am J Phys Med Rehabil 1993; 72: 196–203.
65) Edmonds ME, Jones TC, Saunders WA, Sturrock RD. Autonomic neuropathy in rheumatoid arthritis. Br Med J 1979; 2: 173–5.
66) Gamez-Nava JI, Gonzalez-Lopez L, Ramos-Remus C, Fonseca- Gomez MM, Cardona-Munoz EG, Suarez-Almazor ME. Autonomic dysfunction in patients with systemic lupus erythematosus. J Rheumatol 1998; 25: 1092–6.
67) Huynh C, Ho SL, Fong KY, Cheung RT, Mok CC, Lau CS. Peripheral neuropathy in systemic lupus erythematosus. J Clin Neurophysiol 1999; 16: 164–8.
68) Meyer C, Milat F, McGrath BP, Cameron J, Kotsopoulos D, Teede HJ. Vascular dysfunction and autonomic neuropathy in type 2 diabetes. Diabet Med 2004; 21: 746–51.
69) Straub RH, Antoniou E, Zeuner M, Gross V, Scholmerich J, Andus T. Association of autonomic nervous hyperreflexia and systemic inflammation in patients with Crohn’s disease and ulcerative colitis. J Neuroimmunol 1997; 80: 149–57.
70 Ellenby MS, McNames J, Lai S et al. Uncoupling and recoupling of autonomic regulation of the heart beat in pediatric septic shock. Shock 2001; 16: 274–7.
71) Munford RS, Tracey KJ. Is severe sepsis a neuroendocrine disease? Mol Med 2002; 8: 437–42.
72) Landry DW, Levin HR, Gallant EM et al. Vasopressin pressor hypersensitivity in vasodilatory septic shock. Crit Care Med 1997; 25: 1279–82.
73) Sprung CL, Peduzzi PN, Shatney CH et al. Impact of encephalopathy on mortality in the sepsis syndrome. The Veterans Administration Systemic Sepsis Cooperative Study Group. Crit Care Med 1990; 18: 801–6.
74) Arons MM, Wheeler AP, Bernard GR et al. Effects of ibuprofen on the physiology and survival of hypothermic sepsis. Ibuprofen in Sepsis Study Group. Crit Care Med 1999; 27: 699–707.
75) Yien HW, Hseu SS, Lee LC, Kuo TB, Lee TY, Chan SH. Spectral analysis of systemic arterial pressure and heart rate signals as a prognostic tool for the prediction of patient outcome in the intensive care unit. Crit Care Med 1997; 25: 258–66. |